
Introduction
Sudden cardiac death (SCD), based on sudden cardiac ejection cessation, is an unexpected death occurring within the first hour after symptom onset [1]. SCD accounts for 15–20% of unnatural deaths in developed countries [2]. In China, the incidence of SCD is approximately 4.18/10,000 [3]. Therefore, SCD is a major public health problem worldwide. The prominent symptoms of SCD are chest pain, dyspnea, palpitations, presyncope, and syncope. Primary electrical disorders (PEDs), primary cardiomyopathies, congenital cardiovascular diseases, and primary atherosclerosis are the main causes of SCD [4]. SCD in young individuals (< 35 years of age) is often caused by PED [5].
PED is characterized by cardiac arrhythmias in the absence of detectable structural heart disease [6]. PED encompasses diverse inherited syndromes, predominantly, early repolarization syndrome (ERS), long QT syndrome (LQTS), short QT syndrome (SQTS), Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and arrhythmogenic right ventricular cardiomyopathy (ARVC) [6, 7]. Cardiac muscle contraction and relaxation are triggered by an action potential (AP), which is generated by ionic changes across the cell membrane [8]. Polarization, depolarization, and repolarization of APs result from synergistic activation and inactivation of several voltage-dependent ion channels, including sodium, potassium, and calcium channels [5]. Molecular defects, primarily in these channels, can lead to PED. Since George et al. [9] identified the first ion channel gene causative for malignant arrhythmias, SCN5A, hundreds of arrhythmia-causative/candidate genes have been reported, such as SCN3B (encoding sodium voltage-gated channel beta subunit 3), KCNE1 (encoding potassium voltage-gated channel subfamily E regulatory subunit 1), and RYR2 (encoding ryanodine receptor 2) [9–13].
In this study, we examined 48 Chinese patients with arrhythmia or SCD, who experienced their first seizure under 25 years of age, and identified five mutations in five participants through target sequencing. Of these mutations, the RYR2 mutation (NM_001035.2: c.12269C>T, p.P4090L) and TRPM4 mutation (NM_017636.3: c.2985_3012del, p.E996Gfs*118) are rare, whereas the KCNE1 mutation (NM_000219.5: c.169T>C, p.F57L), KCNQ1 mutation (NM_000218.2: c.853A>C, p.K285Q), and KCNH2 mutation (NM_000238.3: c.793T>C, p.C265R) are novel.
Materials and Methods
Patients and Participants
Between 2017 and 2018, we recruited 48 patients with arrhythmia/SCD across China, with an age of onset under 25 years, to the Department of Cardiology of the Second Xiangya Hospital of Central South University. Patients with Wolff-Parkinson-White syndrome, as well as those experiencing arrhythmias caused by medication, inflammation, or other cardiovascular diseases, were excluded from the study cohort. The Review Board of the Second Xiangya Hospital of Central South University approved this research. Written informed consent was obtained from the patients and their guardians, and all participants consented to participate in this study and to the publication of the images. Blood was collected from patients and their family members.
Target Sequencing
Genomic DNA was extracted with a DNeasy Blood and Tissue Kit (Qiagen, Valencia, Calif., USA). Target sequencing analysis was performed in all patients. A panel of 229 genes (Table S1) including known arrhythmia disease-associated genes were captured with a SureSelectXT2 Target Enrichment System (Agilent, Santa Clara, CA, USA) according to previously reported methods. After enrichment, libraries were sequenced with a HiSeq X-10 system (Illumina, San Diego, CA, USA). All variants were analyzed in SureCall software (Agilent, Santa Clara, CA, USA). Variants with a mean coverage ≥100 were retained. Data analysis followed the methods of Jin et al. [14, 15]. After filtering of common variants (frequency ≥0.01) with the 1000 Genomes Project database (https://www.genome.gov/27528684/1000-genomes-project/), the Chinese Million Variant Database (CMDB, http://cmdb.bgi.com/cmdb/), and the Genome Aggregation Database (GnomAD, http://gnomad.broadinstitule.org), we detected unique single-nucleotide polymorphisms in participants. Subsequently, these variants were predicted by bioinformatics programs including MutationTaster (http://www.mutationtaster.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://provean.jcvi.org/index.php), and Mupro (https://www.ics.uci.edu/~baldig/mutation.html). Conservation analysis was performed in ConSurf Server software (http://consurf.tau.ac.il/). Tolerance analysis was performed in MetaDome software (https://stuart.radboudumc.nl/metadome/dashboard). Gene function, inheritance pattern, clinical phenotype, and pathogenicity were annotated according to the Online Mendelian Inheritance in Man (OMIM) (https://www.omim.org) and American College of Medical Genetics (ACMG) classification [16].
Sanger Sequencing
Cosegregation analysis was performed with Sanger sequencing. Primer pairs were designed in DNASTAR (primer sequences available upon request; Table S2). The target fragments were amplified with polymerase chain reaction and analyzed with an ABI 3100 Genetic Analyzer (ABI, Foster City, CA, USA).
Mutant Modeling
Protein structures were obtained from the Protein Data Bank in Europe database (https://www.ebi.ac.uk/pdbe/?tdsourcetag=s_pcqq_aiomsg). PyMOL was used to build the mutant model according to the wild-type structures.
Results
In total, 48 patients with arrhythmia/SCD with onset under 25 years of age were recruited (Table 1). Among these participants, patient 1 was the youngest and had the most serious case (Table 2). He had recurrent syncope for 3 years and was diagnosed with sick sinus syndrome and cardiomyopathy. Electrocardiography and color ultrasound indicated heart enlargement (Figure 1A, B, Figure S1 and Table S3). Through target sequencing, a set of eight variants in seven genes was identified in this patient (Table S4). Analysis of the bioinformatics predictions, American College of Medical Genetics (ACMG) classification, and genotype-phenotype co-segregation (given that his parents and sister [I:1, I:2 and II:2] did not bear the RYR2 mutation, thus suggesting that his mutation was de novo), we considered that the mutation of RYR2 (NM_001035.2: c.12269C>T, p.P4090L) was disease causing (Figure 1C and Table 3). Alignment of RYR2 amino acid sequences revealed that P4090 is conserved among several species. Similarly, the ConSurf Server software predicted that the P4090 amino acid is located in the conserved region of the RYR2 protein (Figure 2A and B). In addition, MetaDome software predicted that the P4090 amino acid is located in the intolerant region of the RYR2 protein (Figure 2C). The ‘intolerant region’ of a protein, where mutations are poorly tolerated, often contains crucial functional motifs, active sites, or structural domains, making it sensitive to mutations. Furthermore, SWISS-MODEL software was used to explore the spatial configuration of the mutation. The p.P4090L mutation increases the hydrophobic surface area of the RYR2 protein and may cause differences in size, proline residues, and the aliphatic index with respect to those of the WT protein (Figure 3A).
Characteristics and Clinical Phenotypes of all Participants.
| LQTS | SQTS | BrS | ERS | CPVT | SCD | Other arrhythmias* | |
|---|---|---|---|---|---|---|---|
| Age (year) | 18.51 ± 4.27 | 21.25 ± 2.75 | 20.25 ± 2.29 | 16.67 ± 3.18 | 19.43 ± 5.07 | 30.50 ± 4.43 | 25.11 ± 2.48 |
| Onset age (year) | 13.98 ± 2.35 | 17.00 ± 3.34 | 19.25 ± 2.39 | 15.00 ± 3.00 | 14.80 ± 1.54 | 17.67 ± 2.06 | 17.17 ± 1.04 |
| Sex | 3 M, 4 F | 2 M, 2 F | 2 M, 2 F | 2 M, 1 F | 3 M, 2 F | 5 M, 1 F | 13 M, 6 F |
| Number | 7 | 4 | 4 | 3 | 5 | 6 | 19 |
| Proportion | 14.58% | 8.33% | 8.33% | 6.25% | 10.42% | 12.50% | 39.58% |
LQTS, long QT syndrome; SQTS, short QT syndrome; BrS, Brugada syndrome; ERS, early repolarization syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; SCD, sudden cardiac death; M, male; F, female.
*Including sinus arrhythmia, paroxysmal supraventricular tachycardia, ventricular fibrillation, and atrial fibrillation.
Five Patients and their Symptoms.
| Patient | Sex | Age (year) | Symptom | Electrocardiogram test | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| QRS (ms) | QT/QTcB (ms) | PR (ms) | P (ms) | RR/PP (ms) | P/QRS/T (°) | SV1+RVS (μV) | ||||
| 1 | M | 11 | Cardiac arrhythmia(?), sick sinus syndrome, dyspnea, recurrent syncope for 3 years | 82 | 518/530 | - | - | 1090/1665 | -/-46/43 | Null |
| 2 | M | 13 | Early repolarization syndrome, palpitation for 9 months | 90 | 402/421 | 156 | 112 | 902/909 | 32/63/59 | 2665 |
| 3 | F | 14 | Palpitation for 2.5 years, with one episode of syncope | 80 | 418/418 | 182 | 114 | 1002/1000 | 61/53/54 | 3109 |
| 4 | M | 16 | Long QT syndrome, intermittent palpitation for 3 years, syncope | 94 | 450/522 | 212 | 120 | 744/740 | 32/−50/52 | 1420 |
| 5 | M | 35 | Sudden cardiac death | 80 | 400/419 | 134 | 98 | 910/909 | 55/79/44 | 1953 |
M, male; F, female; -, undetected; null, no data; “?”, diagnosis made by another hospital. “_”, data before death.

(A, D, G, J, M) Pedigrees of five patients with segregation analysis. The black symbols represent affected members, and arrows indicate the proband. Genotypes are identified by letters and a slash, with red representing mutations. Question marks represent unknown data or symptoms. (B, E, H, K, N) Baseline 12-lead electrocardiographs of patients 1, 2, 3, 4, and 5, respectively. The results indicated that the patients had arrhythmia. (C, F, I, L, O) Sequencing results of the mutations in RYR2 (c.12269C>T, p.P4090L), KCNE1 (c.169T>C, p.F57L), KCNQ1 (c.853A>C, p.K285Q), KCNH2 (c.793T>C, p.C265R), and TRPM4 (c.2985_3012del, p.E996Gfs*118). The sequence chromatograms indicate heterozygous mutations in every patient.
Mutations in Patients Identified by Target Sequencing.
| Patient | Gene | Mutation | Mutation Taster | PolyPhen-2 | SIFT | CMDB | GnomAD | OMIM clinical phenotype | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|
| 1 | RYR2 | c.12269C>T, p.P4090L | D | D | D | - | - | AD, arrhythmogenic right ventricular dysplasia 2; AD, ventricular tachycardia, catecholaminergic polymorphic, 1 | Likely pathogenic (PS2, PM1, PM2, PP3) |
| 2 | KCNE1 | c.169T>C, p.F57L | D | D | T | - | - | AD, long QT syndrome 5; AR, Jervell and Lange-Nielsen syndrome 2 | Likely pathogenic (PM1, PM2, PP1, PP3) |
| 3 | KCNQ1 | c.853A>C, p.K285Q | D | D | D | - | - | AD, atrial fibrillation, familial, 3; AD, long QT syndrome 1; AD, short QT syndrome 2; AR, Jervell and Lange-Nielsen syndrome | VUS (PM1, PM2, PP3) |
| 4 | KCNH2 | c.793T>C, p.C265R | D | P | D | - | - | AD, long QT syndrome 2; AD, short QT syndrome 1 | Likely pathogenic (PS2, PM2, PP2) |
| 5 | TRPM4 | c.2985_3012del, p.E996Gfs*118 | D | - | - | - | 0.00016 | AD, erythrokeratodermia veriabilis et progressiva 6; AD, progressive familial heart block, type IB | Likely pathogenic (PVS1, PM4, PP1, PP3) |
D, disease causing; T, tolerated; P, polymorphism; AR, autosomal recessive; AD, autosomal dominant; VUS, variant of unknown significance.

(A, D, G, J) Conservation analysis of mutant amino acid sites of RYR2, KCNE1, KCNQ1, and KCNH2, predicted by ConSurf Server software. (B, E, H, K) Alignment of multiple RYR2, KCNE1, KCNQ1, and KCNH2 protein sequences, respectively, across species. Letters in red show affected sites that are evolutionarily conserved. (C, F, I, L) Tolerance analysis of mutant amino acid sites of RYR2, KCNE1, KCNQ1, and KCNH2, predicted by MetaDome software.

Structure Prediction of Wild Type and Mutant Proteins.
(A–E) Protein models of RYR2, KCNE1, KCNH2, KCNQ1, and TRPM4 with or without mutants, predicted by SWISS-MODEL online software. Black arrows show the changes due to the mutation.
Patients 2, 3, and 4 were adolescents, all of whom experienced arrhythmia and palpitation (Table 2, Figure 1D, E, G, H, J, and K). Patient 2 had ERS, and patients 3 and 4 were diagnosed with LQTS with syncope. After target sequencing, ACMG classification, and Sanger sequencing, we identified three unreported mutations, in KCNE1 (NM_000219.5: c.169T>C, p.F57L), KCNQ1 (NM_000218.2: c.853A>C, p.K285Q), and KCNH2 (NM_000238.3: c.793T>C, p.C265R), in these patients (Table 3, Table S4, Figure 1F, I, and L). Segregation results indicated that the KCNH2 mutation in patient 4 was de novo, whereas the KCNE1 mutation in patient 2 was inherited from his mother (I:2), who presented arrhythmia and recurrent palpitation. Patient 3’s mother (I:2) did not bear the KCNQ1 mutation, whereas the patient’s father had died with SCD (Figure 1D, G and J). Conservation analysis and tolerance analysis were subsequently conducted for these affected amino acid sites (Figure 2D–L). KCNE1 mutant modeling indicated that a phenylalanine was substituted by leucine, and the physicochemical properties (including hydrophobicity, size, and aliphatic and aromatic indexes) were altered (Figure 3B). Compared with the WT KCNH2 protein, the C265R mutant may have differences in hydrophobicity, size, charge, polarity, and the cysteine index (Figure 3C). On the basis of the KCNQ1 model, p.K285Q was found to be adjacent to an α-helix, and the electric charge was altered (Figure 3D). Given that these three mutations were predicted to be highly pathogenic through bioinformatics analysis, and were absent from the 1000G and GnomAD controls, we reasoned that these mutations might have caused the disease.
Patient 5 died with SCD (Table 2). According to the medical records, patient 5 had been diagnosed with PED 20 years prior (Figure 1N). Patient 5’s father (II:1) had died of a heart attack at the age of 42 years, and his grandfather (I:1) died in his thirties (Figure 1M). By sequencing, we detected that patient 5 had a TRPM4 mutation (NM_017636.3: c.2985_3012del, p.E996Gfs*118) (Table 3, Table S4 and Figure 1O). SWISS-MODEL software indicated that this mutation might lead to mistranslation of 118 amino acids after the E996 site, and to premature termination, thus truncating the TRPM4 protein (Figure 3E). Our findings suggested that the TRPM4 mutation was disease causing. For other participants, we have not yet identified their pathogenesis.
Discussion
PED typically refers to malignant arrhythmias without apparent structural cardiac changes that might cause SCD [5]. The most important representatives are LQTS, SQTS, BrS, and CPVT. Other arrhythmias, such as ERS, have emerged as new potential PEDs [17]. A review from the United States has described the prevalence of various PEDs. LQTS occurs in 1 in 2000 people and shows a slight female predominance. BrS occurs in approximately 1 in 10,000 people (or a higher rate in East Asia). CPVT in occurs in approximately 1 in 10,000 people. SQTS is very rare [6]. Herein, we recruited 48 patients with arrhythmias/SCD in China, among whom patients with LQTS accounted for the greatest proportion. In addition, each PED has its own typical mode of presentation and most commonly associated gene. The most common genotypes for LQTS are KCNQ1 and KCNH2. SCN5A is the gene most commonly associated with BrS. Approximately 60% of patients with CPVT have a mutation in the RYR2 gene [6]. However, these features were not reflected in our study findings, possibly because of the limited sample size. We recruited six patients with SCD and 42 patients with early-onset arrhythmia, with an age range from 11 to 51 years. To limit environmental effects, we selected young patients with arrhythmia without other organ defects, and a minority of middle-aged patients. Given that patients with cardiac channelopathy are usually unaware of their disorder, and SCD might be the first presenting symptom, screening infants and children for mutations in PED-associated genes may be necessary to provide intervention or prevention strategies for carriers before their first heart attack [5].
AP is an electrical signal that translates into cardiomyocyte contraction generated by a set of ion movements across the cell membrane. The five genes with mutations identified in this study all participate in AP. KCNQ1, KCNE1, and numerous accessory regulatory molecules form a K+ channel (IKs) complex. KCNH2 encodes potassium voltage-gated channel subfamily H member 2, a component of another K+ channel (IKr) complex [18]. Defects in these proteins may lead to abnormal K+ currents in the repolarization state, thus resulting in PEDs. According to reports in the literature, patients with RYR2, KCNQ1, and KCNH2 gene mutations have heterogeneous phenotypes. Here, we summarized these reported phenotypes according to the HGMD, which may aid in the analysis of genotype-phenotype correlations (Figure 4A–C). In contrast to the RYR2 gene, KCNQ1 gene, and KCNH2 gene, few mutations have been reported in the KCNE1 gene and TRPM4 gene. To date, approximately 57 mutations of the KCNE1 gene and 38 mutations of the TRPM4 gene have been identified in patients with PED or SCD (Figure 4D and E). All five mutations reported in this study are rare. The RYR2 mutation (NM_001035.2: c.12269C>T, p.P4090L) is included in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). The TRPM4 mutation (NM_017636.3: c.2985_3012del, p.E996Gfs*118) is included in the GnomAD database, with extremely low frequency (Table 3). The remaining three mutations identified in our study – KCNE1 mutation (NM_000219.5: c.169T>C, p.F57L), KCNQ1 mutation (NM_000218.2: c.853A>C, p.K285Q), and KCNH2 mutation (NM_000238.3: c.793T>C, p.C265R) – were previously unreported and therefore are considered novel. Our study thus expands the pathogenic mutant spectrum of these genes.
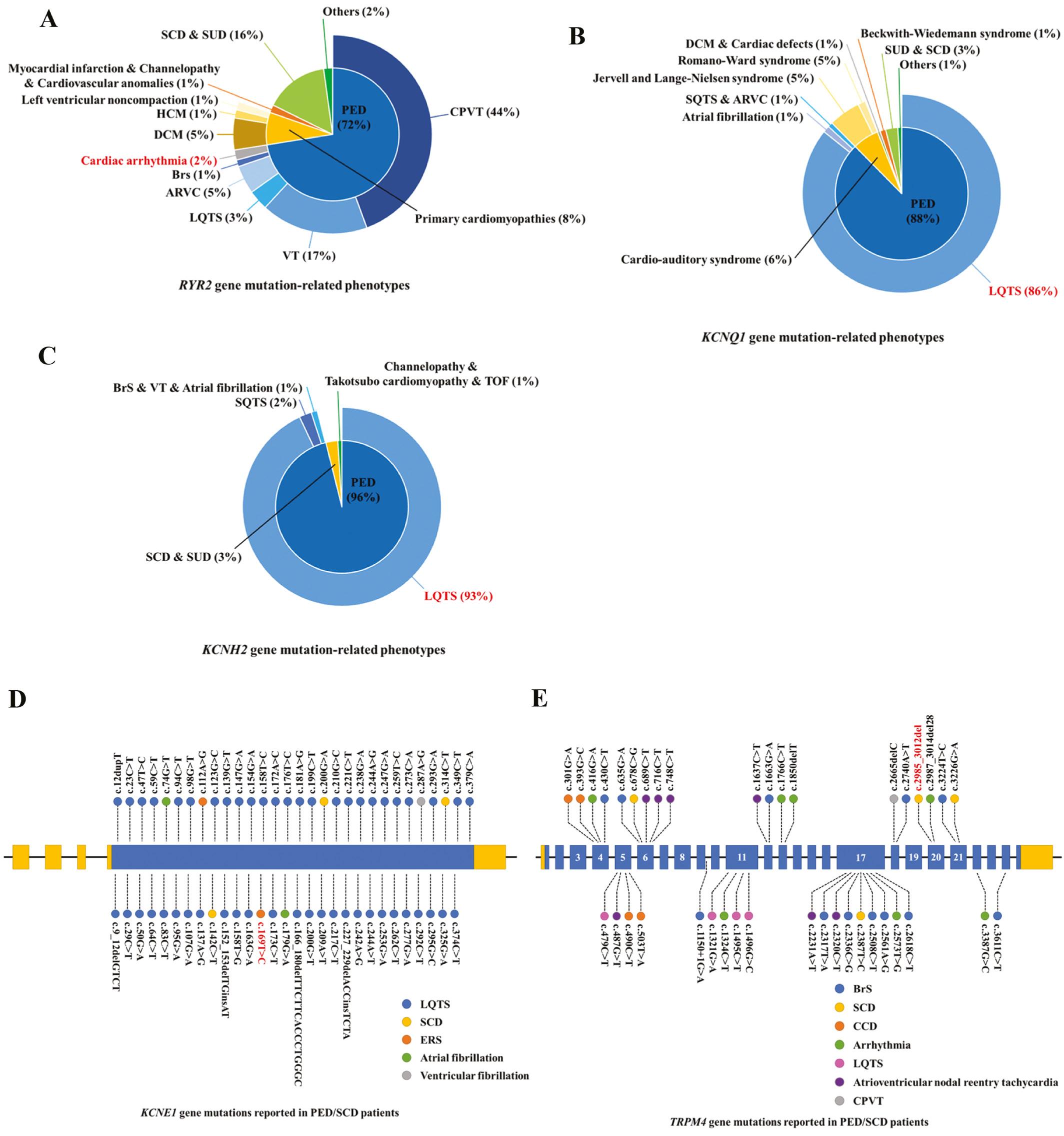
(A–C) Overview of all reported phenotypes caused by RYR2, KCNQ1, and KCNH2 gene mutations. (D, E) Overview of all reported PED or SCD cases caused by KCNE1 and TRPM4 gene mutations. LQTS, long QT syndrome; SQTS, short QT syndrome; BrS, Brugada syndrome; ERS, early repolarization syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; SCD, sudden cardiac death; SUD, sudden unexplained death; VT, ventricular tachycardia; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; TOF, Takotsubo cardiomyopathy.
KCNE1 is an ancillary protein of IKs, which modulates the gating kinetics and enhances the stability of the channel complex [19]. KCNE1 F57 is located in the transmembrane domain, near T58 and L59, which interact with KCNQ1 F339 and F340 [20]. The KCNE1 mutation (c.169T>C, p.F57L) leads to a phenylalanine substitution by leucine; consequently, loss of the benzene ring might affect the spatial structure and physicochemical properties of this region, in agreement with the mutation model, and impair KCNQ1-KCNE1 co-assembly (Figure 3B). Whereas most known KCNE1 mutations cause LQTS [21], our patient and her mother exhibited early repolarization; this finding might be attributable to interindividual differences (Figure 4D). Indeed, KCNE1 plays an important role in repolarization. Although KCNE1 is a classic pathogenic gene for PED, the association between KCNE1 and ERS requires further validation. Our study offers a new perspective for exploring this correlation.
KCNQ1 encodes potassium voltage-gated channel subfamily Q member 1 [22]. KCNQ1 is also highly associated with LQTS, and our patient with a KCNQ1 mutation (patient 3) was suspected to have LQTS. KCNQ1 contains six α-helical transmembrane segments (S1–S6), and K285 sits in the S4–S5 linker, a portion of the channel that links the gate to the voltage sensor [18]. The S4–S5 linker is a mutation hotspot. Similarly to the many known mutations in this region, the KCNQ1 mutation (c.853A>C, p.K285Q) might affect protein structure and function [20, 22, 23]. Protein modeling indicated that the mutant site is located on the edge of an α-helix, and Mupro predicted that K285Q decreases the protein’s stability (Figure 3D). Although the mutation was classified as VUS (Table 3), our analysis results suggest that it still carries probability of being a pathogenic mutation within this family. Thus, we suggest that the KCNQ1 mutation caused patient 3’s symptoms.
KCNH2 belongs to the ether-a-go-go (EAG) family [24, 25]. Most patients with KCNH2 mutations were diagnosed with LQTS, and several cases had SQTS or other arrhythmias (Figure 4C). Patient 4, who carried a KCNH2 mutation, had LQTS and syncope. The KCNH2 mutation p.C265R was located in the proximal NH2-terminal domain. Mutations near C265, such as D259N, R269W, and R273Q, have been identified in patients with arrhythmia [20, 26, 27]. We speculated that C265R might also be pathogenic.
TRPM4 encodes a calcium-activated nonselective ion channel that mediates the transport of monovalent cations across membranes, thereby depolarizing the cell membrane. TRPM4 activity increases with increasing intracellular Ca2+ concentration, but this channel does not transport Ca2+ [28]. TRPM4 is associated with isolated malignant arrhythmias, including sinus node dysfunction, and patient 5 was diagnosed with sinus arrhythmia during his lifetime [29]. In patient 5, the TRPM4 frameshift mutation (c.2985_3012del, p.E996Gfs*118) created a transcript with a premature stop codon, which completely altered the S6 and subsequent domains (Figure 3E). This mutation might potentially disrupt the structure and function of TRPM4, thereby leading to arrhythmias. Because the patient’s father (II:1) died of a heart attack, we did not test his genotype. Similarly, some literature has reported an association between loss-of-function mutations in TRPM4 and PEDs/SCD [30]. Notably, Liu et al. have detected the p.Lys914* mutation in the TRPM4 gene in patients with BrS. Functional and biochemical studies have revealed that this mutation results in decreased expression of the TRPM4 channel [31]. We speculate that the p.E996Gfs*118 mutation in our study might involve a similar mechanism; however, validation would require functional experiments.
Unlike the previously described membrane channel protein genes, RYR2 encodes a ryanodine receptor that is found in cardiac muscle sarcoplasmic reticulum and is a Ca2+ channel component [13]. This channel supplies Ca2+ to the cardiac muscle cytosol from the sarcoplasmic reticulum, not from extracellular fluid, and its dysfunction substantially influences myocardial contraction and cardiac rhythm [4]. In this study, patient 1 carrying the RYR2 mutation (c.12269C>T, p.P4090L) not only suffered from severe arrhythmias but also had cardiomyopathy and sick sinus syndrome. Though not abundant, there are still literature reports linking RYR2 mutations to cardiomyopathy [32, 33]. Given that the voltage clock and Ca2+ clocks (rhythmic spontaneous sarcoplasmic reticulum Ca2+ release) jointly regulate sinoatrial node (SAN) automaticity, and RYR2 is down-regulated in canine SAN dysfunction models, we hypothesized that RYR2 might be associated with sick sinus syndrome [34]. Similarly, a patient with RYR2 -associated myopathy patient has been reported to have SAN dysfunction [35]. The p.P4090L mutation affects a channel region of RYR2, which is a mutation hotspot. Known adjacent mutations (such as p.A4091V and p.A4091T) have been reported in patients with arrhythmia [36]. Thus, we reasoned that the RYR2 mutation (c.12269C>T, p.P4090L) might have been responsible for the genetic etiology in patient 1, although he did not have CPVT or arrhythmogenic right ventricular dysplasia.
We conducted genetic screening on 48 patients through targeted sequencing. On the basis of the sequencing results, we provided comprehensive genetic counseling to the families identified with mutations. We also advised carriers of mutations within these families to undergo regular health check-ups. Currently, only a subset of patients have been found to have gene mutations, and most still have undetected genetic alterations. This finding suggests the possibility of other pathogenic genes beyond our current gene list. In future research, we plan to further update and expand our gene list according to the most recent literature, while improving and refining our existing screening strategies. This process will enable more comprehensive genetic screening of patients who have not yet been identified to carry mutations, thus clarifying their genetic pathogenic factors.
Conclusions
We detected five mutations in Chinese patients with PED or SCD (RYR2: c.12269C>T, p.P4090L; KCNE1: c.169T>C, p.F57L; KCNQ1: c.853A>C, p.K285Q; KCNH2: c.793T>C, p.C265R, and TRPM4: c.2985_3012del, p.E996Gfs*118). This work expands the mutation spectrum of PED-associated genes and may contribute to the clinical diagnosis of PED.